A PBGL Snakemake Workflow and Quatitative Trait Locus on Bulk Segregant Analysis
Software Prerequisites
#sra-toolkit #Download git repository:
#Clone pbgl DNA Proto Workflow
git clone https://github.com/pbgl/dna-proto-workflow.git
#Change Directory to root or project
cd dna-proto-workflow
#Create virtual environment
Mamba env create --file env/all-dependencies.yml
#Activate the environment
conda activate dna-proto
#Make a new directory FASTQ
mkdir FASTQ
#Change directory to FASTQ
cd FASTQ
#Download data from NCBI
#Start with Extremely Tolerant Rice Pool ("Mutant")
wget https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR834931/SRR834931
#Use sra-toolkit to split SRA into foward and reverse FASTQ reads and compress the files
fastq-dump --gzip --split-3
#Rename foward and reverse read files
ET_385_1.fq.gz
ET_385_2.fq.gz
#Now get Extremely Sensitive Rice Pool ("Wild-Type")
wget https://sra-pub-run-odp.s3.amazonaws.com/sra/SRR834927/SRR834927
#Use sra-toolkit to split SRA into foward and reverse FASTQ reads and compress the files
fastq-dump --gzip --split-3
#Rename foward and reverse read files
ES_430_1.fq.gz
ES_430_2.fq.gz
#Follow technical documentation for the DNA Prototype Workflow
https://dna-proto-workflow-master.readthedocs.io/en/latest/
#Download Reference Genome and Index it
#Download annotation files (gff, gtf, protein)
#Build a snpEFF configuration file with soft links if necessary or preferred.
#Provide Meta Data information (Contigs of interest, sample sets and definitions)
#Configure config.yml and snpEff.config to match file pathways and names.\
#Test your workflow on a dry-run
snakemake -npr

snakemake --dag -npr -j 1 | dot -Tsvg > dag.svg
 VariantHTML
VariantHTMLsnakemake -j 4 -kpr
#After PBGL Dna prototype pipeline root directory contains 634 items amounting to 271.4 Gigabytes
#There should be a vcf file in output variants final, lets take a look!
bcftools view /home/michael/dna-proto-workflow/output/variants/final/freebayes~bwa~GCF_001433935.1_IRGSP-1.0~all_samples~filtered-strict.vcf.gz | less -S
#Filter variants for biallelic sites only and SNPS types
bcftools view -m2 -M2 -v snps -o freebayes~bwa~GCF_001433935.1_IRGSP-1.0~all_samples~filtered-strict.Biallelic.vcf.gz freebayes~bwa~GCF_001433935.1_IRGSP-1.0~all_samples~filtered-strict.vcf.gz
#Follow pbgl online documentation
https://github.com/pbgl/QTLseqr
conda deactivate dna-proto
#Create and activate new R Environment
mamba env create --file env/R.yaml
#Set samples
HighBulk <- "ET_385" # SRA-run: SRR834931
LowBulk <- "ES_430" # SRA-run: SRR834927
#set file name of the VCF file to load
file <- "freebayes~bwa~GCF_001433935.1_IRGSP-1.0~all_samples~filtered-strict~snpEff.Biallelic.vcf.gz"
#Specify which chromosomes should be included in the analysis (i.e., exclude smaller contigs)
Chroms <- c("NC_029256.1",
"NC_029257.1",
"NC_029258.1",
"NC_029259.1",
"NC_029260.1",
"NC_029261.1",
"NC_029262.1",
"NC_029263.1",
"NC_029264.1",
"NC_029265.1",
"NC_029266.1",
"NC_029267.1")
df <-
importFromVCF(
file = file,
highBulk = HighBulk,
lowBulk = LowBulk,
chromList = Chroms
)
df_filt <-
filterSNPs(
SNPset = df,
refAlleleFreq = 0.20,
minTotalDepth = 70,
maxTotalDepth = 200,
minSampleDepth = 30,
depthDifference = 100,
#minGQ = 150,
verbose = TRUE
)
df_filt <-
runGprimeAnalysis(
SNPset = df_filt,
windowSize = 1e6,
outlierFilter = "deltaSNP",
filterThreshold = 0.1
)
df_filt <-
runQTLseqAnalysis(
SNPset = df_filt,
windowSize = 1e6,
popStruc = "F2",
bulkSize = c(385, 430),
replications = 10000,
intervals = c(95, 99)
)
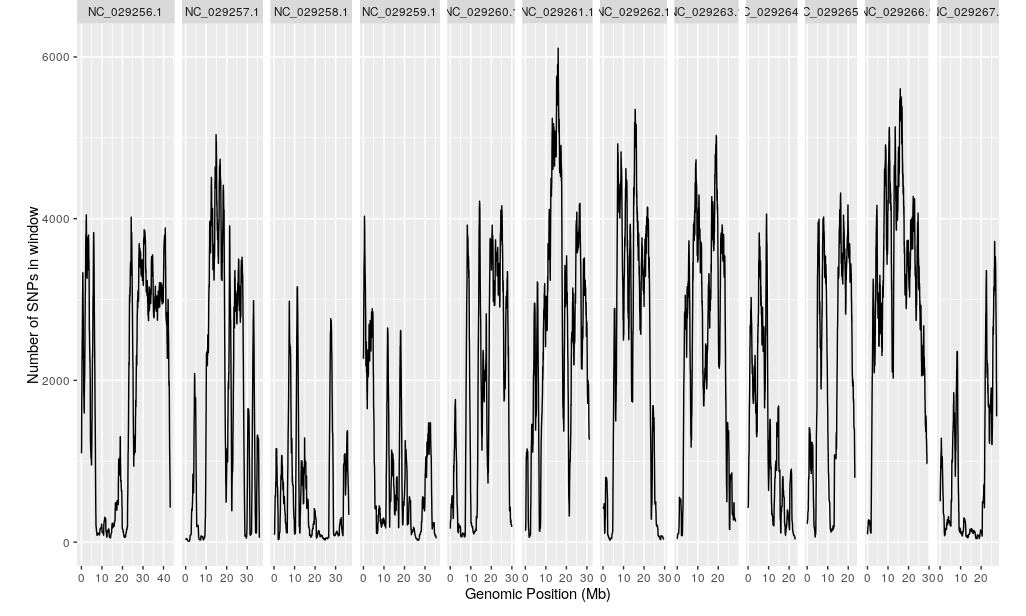
plotQTLStats(SNPset = df_filt, var = "nSNPs", plotIntervals = TRUE)
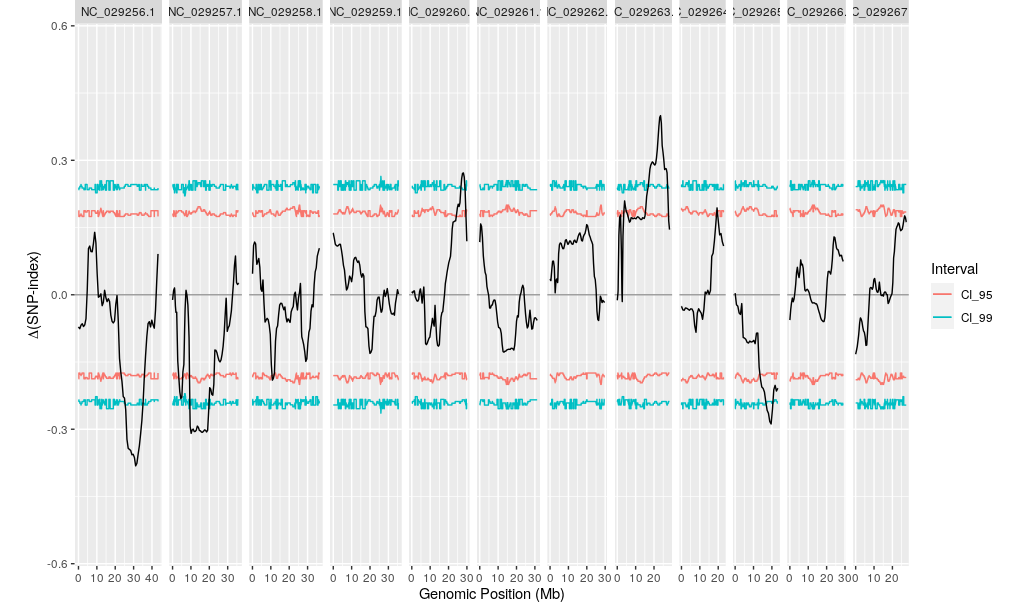
plotQTLStats(SNPset = df_filt, var = "deltaSNP", plotIntervals = TRUE)
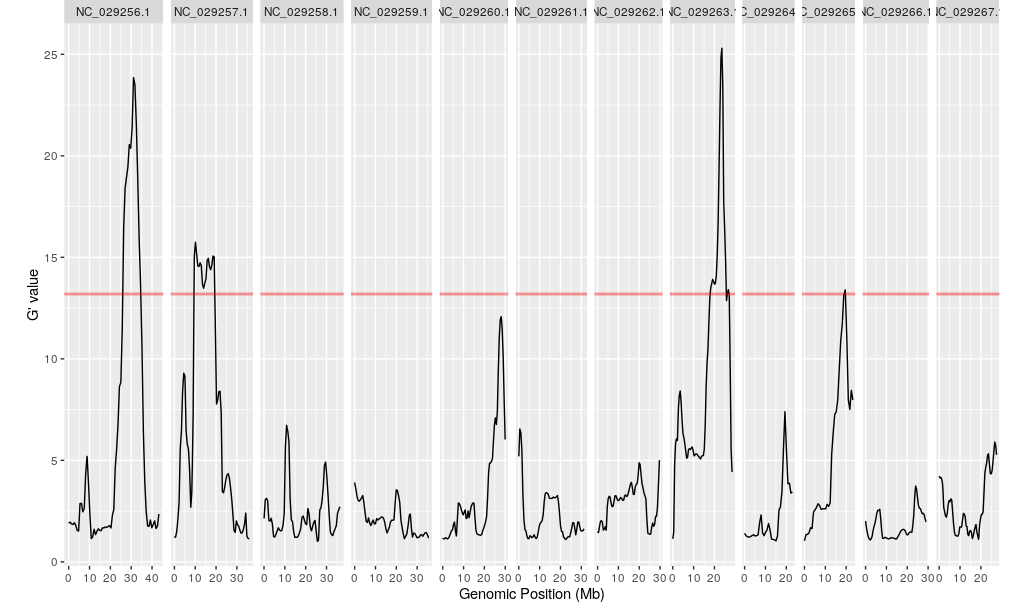
plotQTLStats(SNPset = df_filt, var = "Gprime", plotThreshold = TRUE, q = 0.02)
df_filt$p1 <- df_filt$AD_ALT.LOW/df_filt$DP.LOW
df_filt$p2 <- df_filt$AD_ALT.HIGH/df_filt$DP.HIGH
df_filt3 <- df_filt %>% dplyr::filter(CHROM=="NC_029263.1")
#Perhaps my filter is biased, but here it is. I want observed variants with pvalues below a significant threshold.
df_filt4 <- df_filt3 %>% dplyr::filter(pvalue < .0000962)
write.csv(df_filt3,file = "QTL.csv",sep=",")
bash IGV.sh

Painting The Chromosomes Green
# Clone VCF Hunter from github
git clone https://github.com/SouthGreenPlatform/VcfHunter.git
cd VcfHunter
# Use vcf from Snakemake workflow as in input file to Filter it according to python script vcfFilter.1.0.py
python vcfFilter.1.0.py --vcf freebayes~bwa~GCF_001433935.1_IRGSP-1.0~all_samples~filtered-strict~snpEff.vcf.gz --names Oryza_Satvia_Snakemake/PaintTheChromosomes/all_names.tab --MinCov 10 --MaxCov 300 --MinAl 3 --nMiss 1 --RmAlAlt 1:3:4:5:6 --prefix DNAseq_Filtered -g y
#Use vcftools to split vcf into independent vcf files per chromosome
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029256.1 --recode --out data/Chr01_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029256.1 --recode --out data/Chr01_DNAseq_Filtered.vcf.gz
vcftools --gzvcf Downloads/VcfHunter/bin/DNAseq_Filtered_filt.vcf.gz --chr NC_029257.1 --recode --out data/Chr02_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029258.1 --recode --out data/Chr03_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029259.1 --recode --out data/Chr04_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029260.1 --recode --out data/Chr05_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029261.1 --recode --out data/Chr06_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029262.1 --recode --out data/Chr07_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029263.1 --recode --out data/Chr08_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029264.1 --recode --out data/Chr09_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029265.1 --recode --out data/Chr10_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029266.1 --recode --out data/Chr11_DNAseq_Filtered.vcf.gz
vcftools --gzvcf DNAseq_Filtered_filt.vcf.gz --chr NC_029267.1 --recode --out data/Chr12_DNAseq_Filtered.vcf.gz
#Use python script to produce an allelic frequency plot
python vcf2allPropAndCovByChr.py --conf config/Rice.conf --origin config/RiceOrgin.tab --acc D2_F2_tt --ploidy 2 --dcurve y --col /config/Color.conf --NoMiss --all y
python vcf2allPropAndCovByChr.py --conf config/Rice.conf --origin config/RiceOrigin.tab --acc ES_430,ET_385 --ploidy 2 --NoMiss --all y
#Allelic Frequency Plot (Tolerant Pool ET_385)
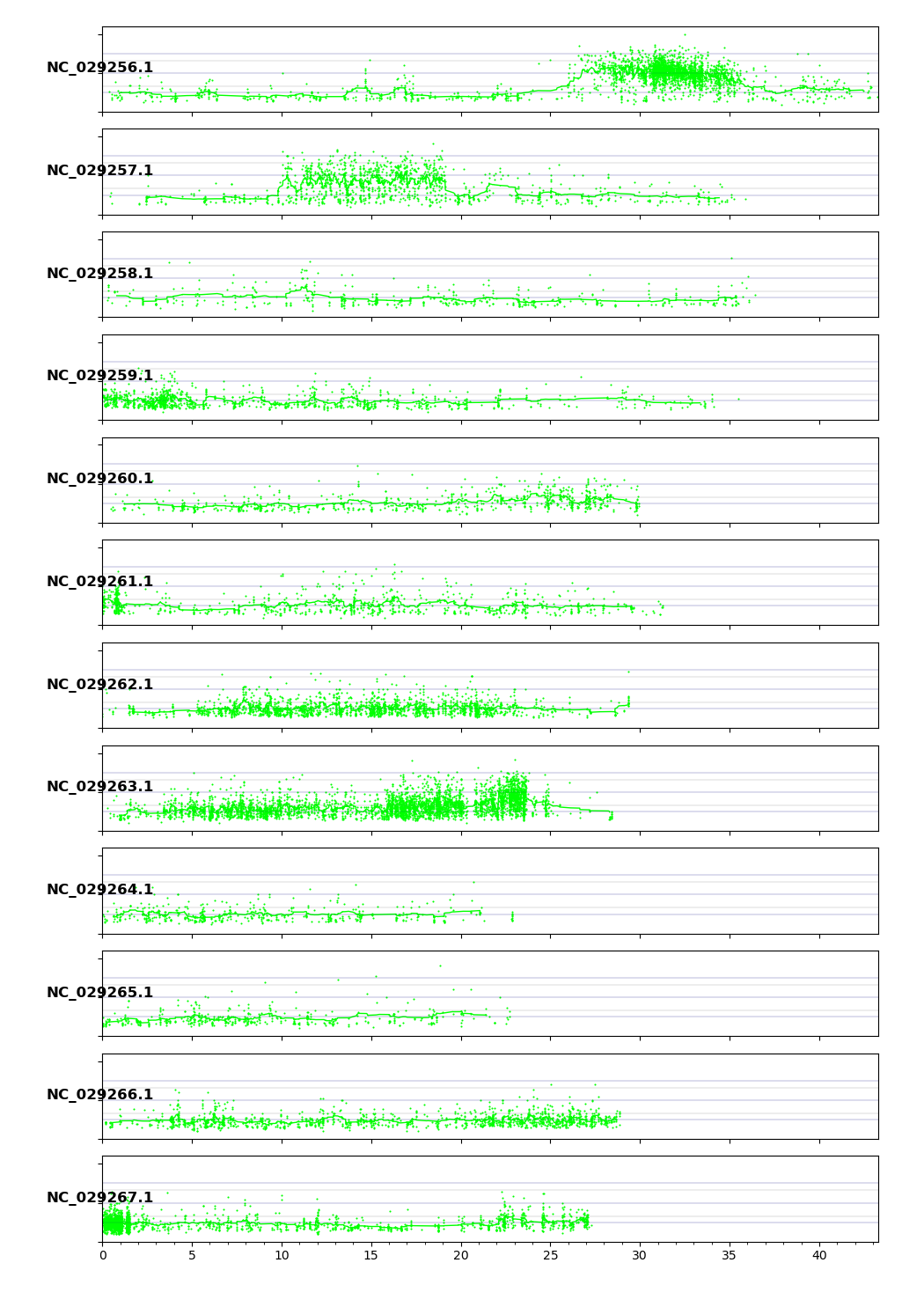
HIGH IMPACT VARIANTS in QTL Region
Download:
SNPEff.latest
https://pcingola.github.io/SnpEff/download/
cd snpEff
cat freebayes~bwa~GCF_001433935.1_IRGSP-1.0~all_samples~filtered-strict~snpEff.vcf | scripts/vcfAnnFirst.py | java -jar SnpSift.jar extractFields - CHROM POS ID REF ALT QUAL FILTER "ANN[*].IMPACT" FORMAT ES_430 ET_385 > impact.tsv
#Import Data into R
All_HIGH_IMPACT <- impact %>% dplyr::filter(ANN....IMPACT == "HIGH")
df_filt2 <- All_HIGH_IMPACT %>% dplyr::filter(CHROM=="NC_029263.1")
#QTL 1 Region on Chromosome 8 from QTL.csv file
df_filt2 <- df_filt2 %>% dplyr::filter(POS >= 17944918 & POS <= 25698533)
df_filt2 <- df_filt2 %>% dplyr::select(POS)
write.table(df_filt2,file="All_High_Impact.txt",row.names=FALSE,col.names=FALSE)
#We know from QTL.csv Chromosome 6 "NC_029263.1" has a QTL peak in the range of positions 17944918 - 25698533!
#Filter original SNP set to include this range of positions on Chromosome 8
df_filt <- df_filt %>% dplyr::filter(CHROM=="NC_029263.1")
#QTL1 Region
df_filt <- df_filt %>% dplyr::filter(POS >= 17944918 & POS <= 25698533)
#Significant SNP
df_filt <- df_filt %>% dplyr::filter(pvalue < .05)
#Write a text file
write.table(df_filt, file = "POSQTL1.txt",col.names = FALSE)
#Use MatchList.py script to find Variants that are
nano MatchList.py
#Make sure file names match what you wrote
#R Script
#df1 <- data.frame(POS1=c(123,457,666,789))
#df2 <- data.frame(POS2=c(123,444,566,789))
#write.table(df1,file="Variant1.txt",col.names = FALSE,row.names = FALSE)
#write.table(df2,file="Variant2.txt",col.names = FALSE,row.names = FALSE)
import pandas as pd
with open("POSQTL1.txt") as file:
list1 = []
for line in file:
list1.append(line.strip())
#print(list1)
with open("All_High_Impact.txt") as file:
list2 = []
for line in file:
list2.append(line.strip())
#print(list2)
found = []
for i in list1:
for j in list2:
if j in i:
found.append(j)
#print(found)
#print(type(found))
found=','.join(found)
#print(found)
#print(type(found))
df = pd.DataFrame([x.split(',') for x in found.split('/n')])
#print(df.transpose())
#print(df)
#print(type(df))
dfT = df.T
df9=dfT.drop_duplicates()
print(df9)
#print(df9['[0]'])
python MatchList.py
18398803
18519533
18593921
19148067
19715648
21045530
21093745
22286217
22636828
23063312
23591197
24582019
24753987
24772963
24775242
24775287
24785765
24854393
#Following protocol from
https://github.com/PBGLMichaelHall/CNVseq
#Remove duplicates from original NCBI fastq foward and reverse Illumina Runs
bash clumpify.sh in=../../../dna-proto-workflow/FASTQ/ES_430_1.fq.gz in2=../../../dna-proto-workflow/FASTQ/ES_430_2.fq.gz out=ES_430.R1.dedup.fq.gz out2=ES_430.R2.dedup.fq.gz dedupe=t -Xmx4096m
bash clumpify.sh in=../../../dna-proto-workflow/FASTQ/ET_385_1.fq.gz in2=../../../dna-proto-workflow/FASTQ/ET_385_2.fq.gz out=ET_385.R1.dedup.fq.gz out2=ET_385.R2.dedup.fq.gz dedupe=t -Xmx4096m
bwa mem -M -t 3 -R '@RG\tID:ES430 \tSM: ES430' ../../dna-proto-workflow/genomes_and_annotations/GCF_001433935.1_IRGSP-1.0/GCF_001433935.1_IRGSP-1.0_genomic.fna ES_430.R1.dedup.fq.gz ES_430.R2.dedup.fq.gz > ES430.dedup.sam
bwa mem -M -t 3 -R '@RG\tID:ET385 \tSM: ET385' ../../dna-proto-workflow/genomes_and_annotations/GCF_001433935.1_IRGSP-1.0/GCF_001433935.1_IRGSP-1.0_genomic.fna ET_385.R1.dedup.fq.gz ET_385.R2.dedup.fq.gz > ET385.dedup.sam
samtools sort -O sam -T sam -T ES430.sort -o ES430_aln.sam ES430.dedup.sam
samtools sort -O sam -T sam -T ET385.sort -o ET385_aln.sam ET385.dedup.sam
python bin-by-sam_2.0.py -o 100Kbin.txt -s 100000 -b -p 2 -c ES430_aln.sam
#Open an R-Studio Session
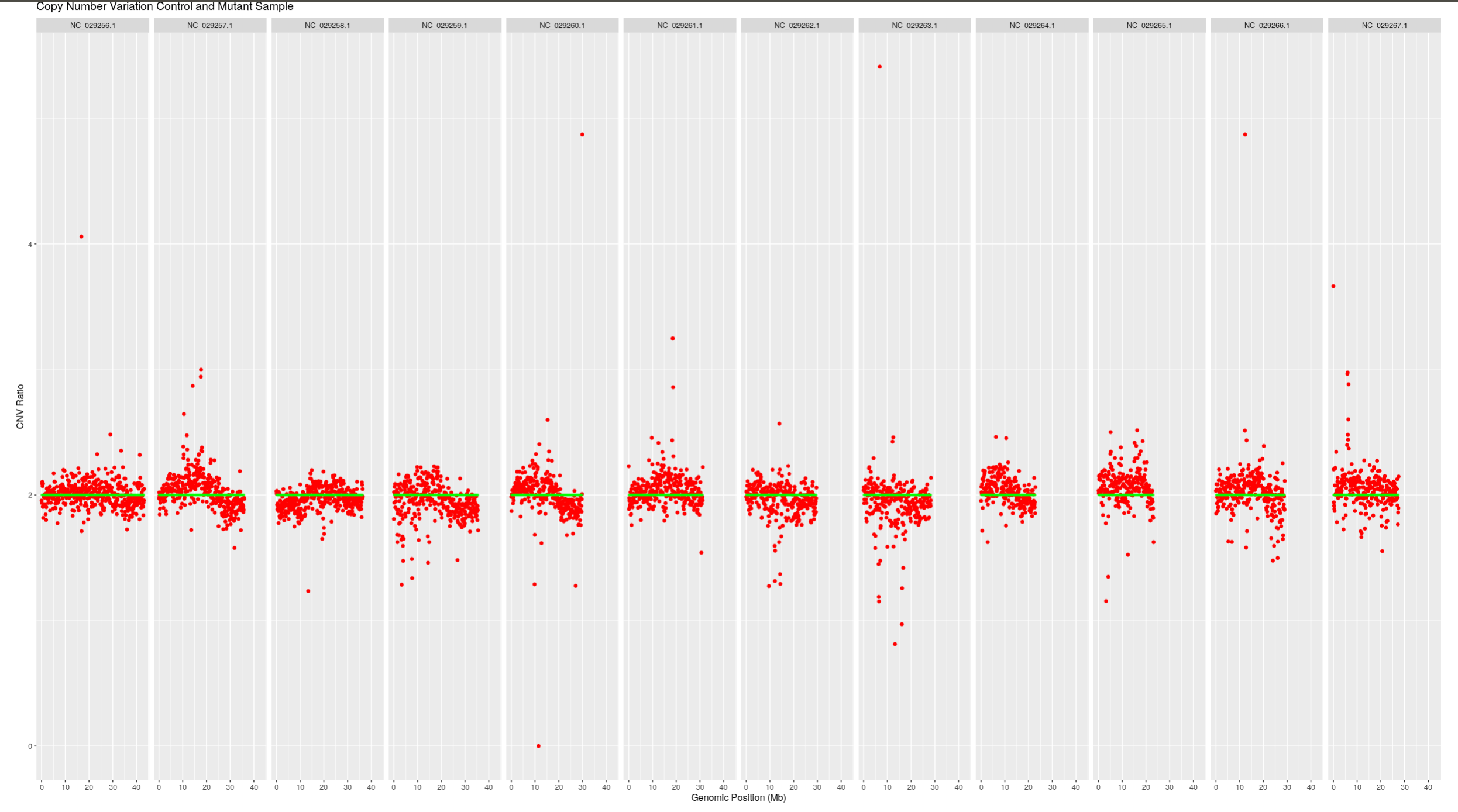
devtools::install_github(repo="PBGLMichaelHall/CNVseq",force=TRUE)
library(CNV)
CNV::CNV(file = "100Kbin.txt", Chromosome = c("NC_029256.1","NC_029257.1","NC_029258.1","NC_029259.1","NC_029260.1","NC_029261.1","NC_029262.1","NC_029263.1","NC_029264.1","NC_029265.1","NC_029266.1","NC_029267.1"),mutantname="ET385.ES430",controlname="ES430.ES430",size=.75,alpha=.25,color="green")